新薬の育薬における役割
新薬の製造販売後に行われる市販後調査を中心とした、新薬を「より使いやすく、より有効性で且つ安全性のある薬に育てる」ことを「育薬」と呼んでいます。
また新薬の治験終了後、国に承認されるまでの過程を「創薬」と呼んでいます。
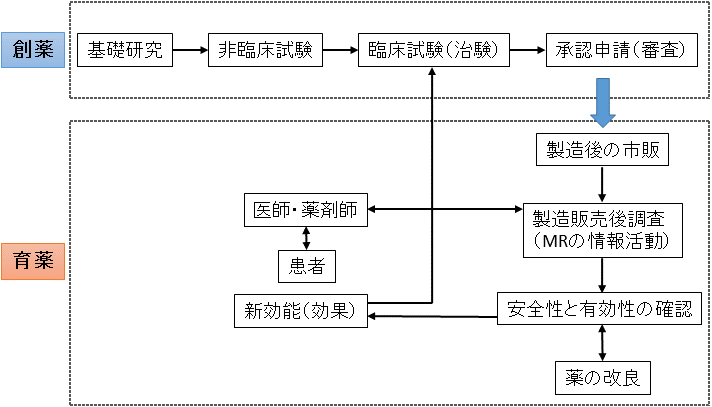
新薬市販後の重要ポイント
代表的な創薬の過程である治験では、下記のような制限があります。
- 高齢者や小児は除外対象のケースが多い。
- 投与期間が短く、実際の効果について検出できないケースが多い。
- 複雑な疾患の患者は、被験者としては除外対象になっている。
- 被験者数が限定されている。
- 併用薬が制限されている。
新薬の有効性と安全性については、承認申請に必要な最低限の情報のみとなりますので、市販後においてもさらに新薬の情報収集の継続を行い、実際の有効性と安全性の確認の継続を行い、より適切な用い方を得ることが育薬であり、育薬において薬の専門家である薬剤師の役割は重要となります。
日本では国民皆保険制度のため、新薬の発売直後には製薬会社により大々的に宣伝が行われ、多くの薬が販売されて使用されるという現実があります。
市販後に多くの患者へ使用されることで、治験では分からない予期していなかった安全性に関する情報が得られることがありますので、薬剤師は常に患者からと製薬会社からの情報についてアンテナを張り、副作用報告などの重要な行動を起こす必要性があります。
医薬関係者からの副作用報告制度
医薬品の副作用または医療機器の不具合で、危害の発生または拡大を防止する必要があると認められた場合、薬剤師はその旨を厚生労働大臣に報告する必要があります。
この薬事法の規定は薬剤師のみならず、 すべての医療機関の開設者、 医師、 歯科医師、登録販売者、 獣医師、 店舗販売業者や医療に携わる者に課せられた義務(薬事法第77条の4の2第2項)となっています。
なお、報告の対象となる症例(情報)については、医薬品または医療機器との因果関係が必ずしも明確でない場合も報告の対象となります。具体的には下記のケースが該当し、 厚生労働省医薬食品局安全対策課が窓口となります。
①死亡
②障害
③死亡につながる恐れのある症例
④障害につながる恐れのある症例
⑤治療の為に病院や診療所の入院、又は入院期間の延長を必要とする症例(③,④の症例を除く)
⑥1~5までに掲げる症例に準じて重篤である症例
⑦後世代における先天性の疾病又は異常
⑧当該医療品又は医療機器の使用によるものと疑われる感染症による症例等の発生
⑨当該医療機器の不具合の発生のうち、①~⑦に掲げる症例等の発生
⑩1~8に示す症例以外で軽微ではなく、且つ添付文書等から予測不能で未知の症例等の発生
⑪当該医療機器の不具合の発生のうち、⑩に掲げる症例の発生の恐れのあるもの
医療機関での医薬情報(DI)活動
病院における入院調剤技術基本料(薬剤管理指導料100点)では、その算定条件となる施設基準に医薬品情報管理室の設置と、薬剤師による医薬情報活動を行うことが明記されています。
多くの医療機関では、薬剤部(薬局)に配置された医薬品情報管理室の担当薬剤師が、製薬会社の医薬情報担当者(MR)との窓口となり、添付文書の改訂などの製薬会社からの情報を整理して医療機関内に伝達しています。
臨床現場で生じた医薬品の有効性・安全性に関する情報の収集と、製薬会社や国への情報伝達は、薬剤師が担う役割となります。
日本における製造販売後の制度
「再審査制度と安全性定期報告」「再評価制度」「副作用・感染症報告制度」という製造販売後の制度があります。
- 医薬品の再審査制度
再審査制度は育薬の中で国が行う重要項目で、新薬の承認の際に再審査対象医薬品と再審査期間を定め、 製薬会社は再審査期間内に再審査に必要な新薬の情報収集を行う必要があります。
- 医薬品の製造販売後の調査及び試験実施の基準(GPSP省令)
「医薬品の臨床試験実施の基準(GCP省令)」に基づいて治験が行われるのと同様に、 製造販売後において再審査のため製薬会社による新薬の情報収集は、「医薬品の製造販売後の調査及び試験実施の基準(GPSP省令)」 に基づいて行われます。
- 安全性定期報告
新医薬品の承認時に厚生労働大臣が指定する期間ごとに、国内における使用状況と副作用情報に、 海外の副作用の発生動向(PSUR)などを盛り込んで、厚労省へ報告するよう製薬会社に義務づけられています。
- 医薬品、医薬部外品、化粧品および医療機器の製造販売後安全管理の基準(GVP省令)
「市販後調査の実施基準」はGPSP省令とGVP省令に分類され、「安全管理情報」は「医薬品等の品質、有効性および安全性に関する情報や医薬品等の適正な使用のための情報」として定義され、安全管理情報の収集、検討、及びその結果に基づく必要な措置は製薬会社に義務づけられています。
またGVP省令で、「医薬情報担当者(MR)」が「医薬品の適正な使用に資するために、医療関係者を訪問すること等により安全管理情報を収集し、提供することを主な業務として行う者」と定義されています。
さらにGVP省令では市販直後調査が定められており、販売開始後に診療における新薬の適正使用を促し、合わせて副作用情報の収集を行うことで、市販直後の治験では予測できなかった副作用にも対応できる仕組みを制定しています。
- 医薬品の再評価制度
現在の医学・薬学の学問的水準に照らして、医薬品の品質や有効性と安全性を見直す仕組みが再評価制度です。再評価については、「定期的再評価」と「臨時の再評価」があります。
定期的再評価
→定められた期間ごとに有効性・安全性について見直しを行う。
臨時の再評価
→緊急な問題発生時や薬効群全体に問題が発生した時に、臨床評価ガイドラインなどにより有効性・安全性の見直しが示唆されたときに行う。
また、薬事・食品衛生審議会にて審議される評価では、内用固形製剤の「品質再評価」があり、溶出試験を中心に製剤の品質を一定水準に保つために行われます。
- 副作用・感染症報告制度
この制度は「企業報告制度」「感染症定期報告制度(生物由来製品)」「WHO国際医薬品モニタリング制度」からなります。
企業報告制度
→薬事法により製薬企業に課せられた義務で、これを怠ると行政処分を受ける制度。
感染症定期報告制度(生物由来製品)
→人や動物に由来する原料で製造される生物由来製品について特別に行われる制度。
WHO国際医薬品モニタリング制度
→国際的な副作用情報の情報交換を行う制度。
- 医薬品副作用の被害救済制度
病院・診療所で処方された医薬品と薬局での医薬品を対象に、使用方法に基づいたにも関わらず副作用により入院、又は入院を必要とする状態で健康を損なう被害に遭った際に、医療費等を給付する制度です。
この制度を利用するには、 副作用による健康被害を受けた本人、又は遺族が投薬証明書と診断書を添付した請求書を、独立行政法人医薬品医療機器総合機構へ提出する必要があるので、薬剤師の協力が必要不可欠となります。
医薬品は承認申請までの治験だけではなく、製造販売後においても有効性・安全性が常に確認され育てられています。この育薬の過程においての薬剤師の役割は、薬の専門家としての重要な責務があり、常に育薬を行っているという自覚に基づく行動が求められます。